
FDA registration of medical devices
Many of you will have heard that FDA approval is necessary to legally sell medical devices in the USA. These procedures include a Premarket Notification 510(k) or a Premarket Approval (PMA). There is also another, lesser-known procedure. The so-called De Novo Program of the FDA. But “only” having product approval is not enough. In addition to the approvals, there are other requirements that must be met: Your quality management system must meet the requirements of the Quality Management System Regulation (QMSR), and products and companies must be registered with the Food and Drug Administration. In this blog, we will explain which steps are necessary on the way to US approval.
FDA approval from the Food And Drug Administration (FDA)
The FDA is a U.S.-based agency. It reports to the Department of Health and Human Services and is responsible for overseeing food, drugs, and medical devices. As is typical for such an agency, its core task is to protect citizens. To fulfill this task, certain devices cannot be sold in the USA without registration. They need FDA registration.
In contrast to authorities in Germany, the FDA has a certain influence on the requirements to be observed within the scope of FDA approval. It has the authority to enact certain laws that must then be observed within the scope of approval. These laws are included in Section 21 of the Code of Federal Regulations (CFR).
The FDA is considered a very strict authority worldwide. This leads to most companies being downright afraid of an FDA audit. Of course, the FDA has the option to order an import ban or similar unpleasant measures. However, a sales stop can also happen to you in the EU if requirements are not met. The key to a good night’s sleep in both the EU and the USA is therefore good preparation and implementation of regulatory requirements.
There are guidelines for many topics that are relevant to FDA approval. These specify the authority’s expectations and actually help you to position yourself well from a regulatory perspective.
Deviations & FDA Approval
Deviations in the FDA registration process can occur during the registration procedure (pre-market) or during an existing registration (post-market).
During the registration process, the FDA will inform you of the respective deviations, and these must be rectified by you within the specified deadline. The deadlines are specified in the respective procedures. Since the FDA’s reviews are very detailed, deviations in registrations are common. The important thing is that the deviations are not so serious that they become a problem in the project. However, you largely rule this out through good preparation and expertise in technical and regulatory aspects. In our projects so far, there have been no deviations so serious that an registration has failed. The deviations occurring during FDA registration are not published.
Deviations that occur with existing FDA approval, i.e. with products on the market, can be published within the scope of a Warning letter. Reportable incidents are published in the USA via the FDA MAUDE database.
Deviations will usually occur in the following stages:
- Level 1: FDA Form 483 (Deviation report by the FDA)
- Level 2: Warning letter
- Stage 3: Sales stop
For example: If a deviation is determined by means of Form 483 and the company does not react sufficiently to it, a warning letter will be published. If there is still insufficient reaction, the FDA will issue a sales stop. During the process, the manufacturer has the opportunity to speak with representatives of the FDA about the procedure and discuss measures. A sales stop, therefore, does not usually come as a surprise.
The publication of deviations can lead to damage to the company’s image. You can view a list of published warning letters from the FDA: Warning Letter .
*21 CFR Part 820 – QSMR
What?! What sounds a bit cryptic here is the reference to the FDA’s Quality Management System Regulation. It describes the most important requirements for a quality management system with which you are allowed to sell products in the USA. It also describes which records the FDA expects from you – so taking a look at these requirements is interesting long before US approval. By switching from QSR to QMSR, the requirements for the quality management system are more closely aligned with EN ISO 13485. In §820.7 “Incorporation by reference.” the QMSR refers to ISO 9000 as the basis for various terms and to ISO 13485 with regard to requirements for the quality management system. This change is triggered by a “Final Rule” issued by the FDA and effective from February 2nd, 2026. At this point, the far-reaching powers of the FDA can also be seen.
It is absolutely welcome that the FDA is orienting itself towards the internationally known and established requirements of ISO 13485. Due to the effectiveness from February 2nd, 2026, it is still necessary to comply with the previous requirements of the QSR until then:
In 21 CFR 820 you will find information on the subject of design controls or the development of medical devices. At this point, it is recommended to compare the requirements of the QSR with your existing development process. It may sound simple, but a manufacturer must implement and maintain procedures for defining and documenting the design output in a way that allows an adequate assessment of conformity with the requirements of the design input. We have often found that it was not entirely clear to customers what the design output of their development process actually is. In the verification, aspects were then partially checked that do not sufficiently prove that the requirements of the design input were implemented in the design output.
With regard to the requirements of the Quality System Regulation, you are already well positioned with an existing QMS according to ISO 13485. However, there are many subtleties that need to be considered. We advise you to use a gap analysis to find out which requirements you have not yet implemented or have not yet implemented sufficiently. Before US approval, make sure that your company is well prepared at the QMS level.
Classification of medical devices for FDA approval
The procedures for FDA approvals are basically related to the product risk. The riskier a product, the higher its risk class. It is important to note that the FDA has its own classifications. The classes from the EU cannot simply be transferred. Incidentally, this also applies to what is classified as a medical device or not.
The FDA recognizes three risk classes:
| Klasse | Beschreibung |
|---|---|
| I | Niedriges Risiko: Zulassung über Listing oder 510(k) |
| II | Mittleres Risiko: Zulassung über Listing oder 510(k) |
| III | Hohes Risiko: Zulassung über PMA (nur selten über 510(k)) |
Use of the "Device Classification Panels" in FDA approval
Most medical devices can be classified by selecting the appropriate category via the Medical Specialty of the Device Classification Panel. The following figure shows this.
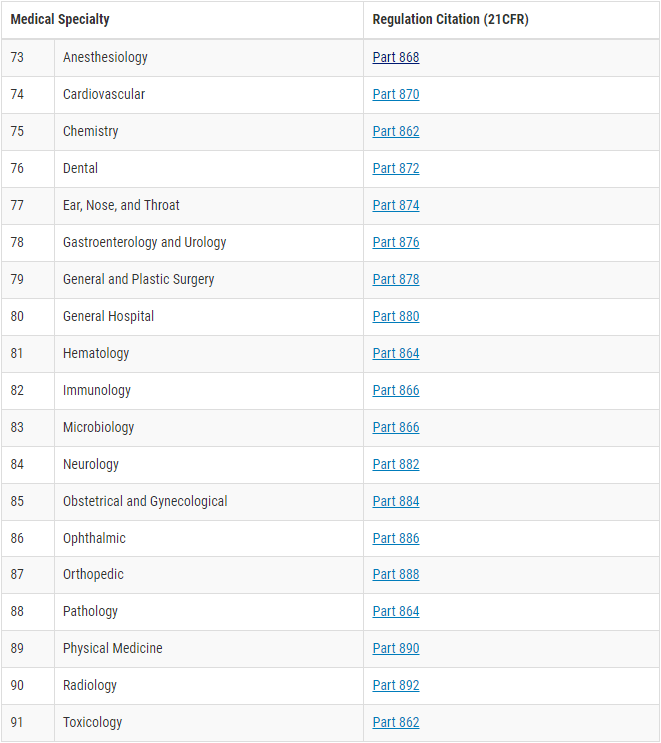
Let’s assume we want to approve a PCR test for the detection of HIV. So we open the Medical Specialty 82 Immunology and look there for the appropriate product type.

A PCR test is a test method based on nucleic acid amplification technology (NAT). The correct entry is therefore “§ 866.3957 – Human immunodeficiency virus (HIV) nucleic acid (NAT) diagnostic and/or supplemental test.”
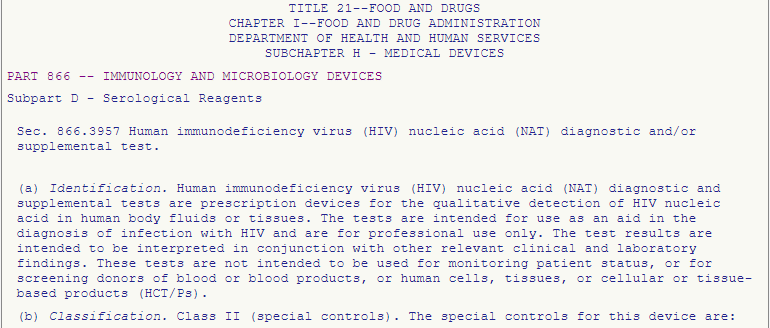
The product is therefore classified in Class II and must meet the special controls listed under § 866.3957.
Determining a suitable FDA approval procedure
We have already described that you must list your products and register your company. However, in order to be able to list products, you must first approve them – if required. If this is not necessary, you also do not need a unique registration number for the product, which you will receive upon successful completion of an approval. You can therefore list the products with the FDA without approval.
But let’s assume that your product must be formally approved and a listing is not sufficient. How do you know whether you should use a Premarket Notification 510(k), a Premarket Approval or the De Novo Program of the FDA?
Do you know the risk class of your product? This is a first important indication for choosing one of the approval procedures. Do you know the Product Code, via which the appropriate product type is regulated by the FDA? That’s ideal! Because this allows you to find out which requirements you have to comply with.
Determining a Product Code for FDA approval
You can find Product Codes using the FDA’s 510(k) Premarket Notification Database or the Premarket Approval (PMA) Database. For example, enter a product from another manufacturer that is similar to your product in the Device Name field of the 510(k) database. In this example, we are using the approximate name of an endoscope from a well-known manufacturer. We have not obtained permission to use the product name and related information. For this reason, the corresponding fields in the graphics are deleted.

After you have entered the approximate Device Name, you will be shown a list of search results. Choose the most appropriate result. You will then be shown the following view, from which you can extract the desired Product Code.
As you can see, this endoscope is even assigned multiple Product Codes. This is related to the intended use and means that you must comply with the requirements for each individual Product Code.

You can now open the codes and the Regulation Number. For the code NWB, for example, the following is displayed:

This overview shows you that you must use a 510 k as the approval type. The recognized consensus standards are standards that you should comply with for products of this code. You can find the legal basis from the Code of Federal Regulations (21 CFR) in the Regulation Number.
In this way, you can determine step by step the requirements for each individual Product Code that is applicable to your product. In the “ideal case”, of course, it’s only one, which makes your life easiest. But as you can see, there are products where the intended use and range of functions lead to multiple codes.
FDA approval: Approval types according to Product Code
- A Premarket Approval (PMA) is usually required for Class III products.
- If you were able to determine a Product Code, a Predicate Device is available, and the Product Codes require a 510(k), you can take the route of a Premarket Notification to get your product approved. The comparison with the Predicate Device is the basis for argumentation in the registration. All differences are discussed and assessed for their impact on safety and performance.
- Couldn’t determine a suitable Product Code for your product? This means your product type hasn’t been approved by the FDA yet. New products like this are initially classified as Class III by the FDA. If you want your product to be classified as Class I or II, the FDA’s De Novo Program is the right approach.
- Your product does not require FDA approval? In this case, you must still list the product with the FDA.
Registration even without FDA approval: "Registration and Listing"
Companies involved in the manufacturing and distribution of medical devices for use in the United States must register annually with the FDA. This process is known as “establishment registration”. In 2020, the cost for this was $5,236. For the fiscal year 2026, which begins on October 1st, 2025, the costs amount to USD 11,423.
Most companies required to register with the FDA must also list their products for the US market. If a product needs Premarket Approval or Premarket Notification before being marketed in the USA, the identification number of the registration must be provided. Registration and listing are done electronically.
U.S. Agent for FDA approval
If your company is considered a “foreign establishment” and is involved in the manufacturing, preparation, distribution, assembly, or processing of a product imported into the USA, you need a U.S. Agent.
The U.S. agent must either be resident in the USA or maintain a branch there. His responsibilities relate to:
- supporting the FDA in communicating with your company.
- answering questions regarding your products that you want to market in the USA.
- supporting the FDA in planning inspections in your company.
- If the FDA is unable to contact your company directly or quickly, the FDA may provide information or documents to the U.S. Agent, and such action will be considered equivalent to providing the same information or documents to your company.
As part of your “facility registration”, you provide the name of the U.S. Agent. This is done electronically through the FDA Unified Registration and Listing System (FURLS system).
FDA approval: important procedures at a glance
FDA Pre-Submission Program
The FDA’s so-called pre-sub is a formal procedure that takes place before the actual approval. It serves to clarify open questions regarding the strategy and usually comes before procedures such as 510(k), De Novo Request or PMA.
Premarket Notification (PMN) or 510(k) approval
The 510(k) approval procedure is not possible for novel products, but is used when there are already comparable approved products. With a 510(k), reference is made to an already approved product for approval, which is particularly helpful with regard to the necessity of clinical data. This enables comparatively fast market access and the 510(k) is to be regarded as the most important procedure for FDA approval of medical devices in the USA.
Several options are available for this process. The traditional 510(k) variant is intended for products with similar predecessor products, whereas the Special 510(k) variant is used for changes to your own products, for which an accelerated procedure is used. Finally, there is the so-called Abbreviated 510(k) procedure, which checks compliance with the “Special Controls” and “Guidance Documents”. The eStar program now offers the possibility of digital FDA approval of 510(k)s
Premarket Approval (PMA)
Novel products or Class III products must go through the PMA approval process, which is typically quite complex and time-consuming and is therefore too complex for manufacturers of actually non-critical products. The reason for the increased requirements: Class III includes high-risk products that have correspondingly high requirements for their approval.
De Novo Classification Request / De Novo Procedure
This is the FDA approval procedure for novel medical devices. This means that it is a product for which there is no product (Predicate Device) approved by the FDA, but which is to be classified in Class I or II. A distinction is made between the classic de novo procedure approach and the direct de novo approach:
The classic approach begins with the 510(k) application, the Premarket Notification. If this is rejected by the FDA because it could be a new product, the manufacturer can submit the direct de novo application, which, however, only proves to be effective if the manufacturer can assume that no comparable product exists and that its own product will probably be classified according to Class I or II.
FDA approval: Summary
There is a lot to consider even before product approval in the USA:
- Your quality management system must meet the requirements of the Quality System Regulation 21 CFR Part 820 and, if applicable, other regulatory requirements.
- It should be noted that a changeover to the Quality Management System Regulation is taking place and thus an approximation to ISO 13485.
- You must register your company with the FDA and appoint a U.S. Agent.
- Then, taking into account the risk class and the Product Code, it is important to identify the correct approval procedure and the requirements, standards and guidances applicable to the product.
- Only then does product approval begin via a Premarket Notification 510(k), Premarket Approval or as De Novo.
Professional help with FDA approval for medical devices
If you need help with FDA approval, we at thinqbetter are definitely the right partner for you.
- Our qualification – Through the support of more than 140 different companies, we can bring extensive experience in various specialist areas to the table. In addition, we are constantly working on optimizing and improving our work processes, project durations and quality. Our team of specialists from various natural sciences (e.g. medical technology, biology, chemistry, biotechnology, immunology) is also regularly trained to be optimally equipped for all services offered at all times.
- Our values – We act reliably and seriously and enjoy the trust of many long-standing customers. Medical technology is strictly regulated and the large number of different requirements poses a major challenge for manufacturers. That is why we are always at your side as a reliable, competent and trustworthy partner – both as a consultant and as a service provider. We are committed to ensuring that your products reliably obtain FDA approval
- Our range of services in the field of FDA approval – We have tailored our B2B offers for manufacturers of medical devices and manufacturers of in-vitro diagnostics (IVDs). In addition to a free initial consultation, we can offer you the following services in the field of FDA approval:
- Creation or compilation or support in the creation of Premarket Notifications, also called Premarket Notifications 510(k) / 510(k) / PMN / PMN 510(k).
- Creation / compilation or support in the creation of Premarket Approvals
- Creation / compilation or support in the creation of De Novo Classification Requests
- Creation / compilation or support in the creation of Presubmissions before FDA approval
- Support with Facility Registration and Device Listing with the FDA
In addition, there are other services and advice on regulatory topics.
If you need support with the upcoming FDA approval it is best to contact us right away. This gives us enough time to take all the necessary measures and guide you safely through the FDA approval !
Frequently asked questions
What is the Significance of the Warning Letter in FDA Approval?
If a significant violation of the requirements is determined with existing FDA approval, the FDA can formally inform the manufacturer about this in the form of a Warning Letter. This may include, for example, failure to meet necessary requirements for production processes or inadmissible advertising statements. In this case, appropriate corrective measures must be initiated immediately. A corrective action plan must therefore be submitted within 15 days. Failure to react could lead to criminal prosecution in the worst case. A Warning Letter is published by the FDA and is accessible to everyone.
What are the Important Prerequisites for FDA Approval?
The term “approval” is mostly used for several things, although it is not always the same thing. The term FDA approval is mostly used as a synonym for the fact that a medical device has gone through any procedure that allows it to be sold in the USA. The FDA itself speaks of a so-called “clearance” in the case of a 510(k). Terms such as “FDA Approval” should be avoided. In any case, the respective legal requirements for the product and the quality management system must be met.
What are the Differences between FDA Approval and EU Approval?
Both the USA and the EU want to ensure the conformity of medical devices. Although the goals are practically the same, there are, for example, differences between the product classifications and approval procedures. One of the main differences is the possibility of carrying out a Premarket Notification 510(k) without clinical data, even if a clinical trial would be necessary in the EU, for example. Another difference lies in decentralized approval procedures by notified bodies compared to the central, official FDA approval.